




 收藏
收藏
| 规格 | 价格 | 期货 | 数量 |
|---|
| 项目 | 性能 |
| 基质 | 4%琼脂糖凝胶 |
| 载量 | >20 mg 6×his-tagged protein/ml 基质 |
| 微球粒径 (μm) | 45-165 |
| 最大压力 | 0.1 MPa, 1 bar |
| 储存缓冲液 | 含20% 乙醇的1×PBS |
| 储存温度 | 2-8℃ |
| 试剂种类 | 浓度 |
| 还原剂 | 10 mM β-mercaptoethanol1 |
| 变性剂 | 8 M urea 6 M Gu-HCl |
| 去污剂 | <1% Triton™ X-100 1% NP-40 1% CHAPS,SDS,sarcosyl |
| 其他类 | ≤500 mM imidazole2 at pH 7.0 to 8.0 for elution 30% ethanol3 20% glycerol 500 mM KCl 1.0 M NaCl 20 mM MES 50 mM Tris4 50 mM HEPES 50 mM MOPS |
| 名称 | 体积 | 配方 |
| Lysis Buffer |
1L | 50 mM NaH2PO4 (7.80 g NaH2PO4·2H2O) 300 mM NaCl (17.54 g NaCl) 10 mM imidazole (0.68 g imidazole) 使用NaOH溶液调节pH至8.0, 使用0.22 或者0.45 μm滤膜过滤除菌。 |
| Wash Buffer |
1L | 50 mM NaH2PO4 (7.80 g NaH2PO4·2H2O) 300 mM NaCl (17.54 g NaCl) 20 mM imidazole(1.36 g imidazole) 使用NaOH溶液调节pH至8.0, 使用0.22 或者0.45 μm滤膜过滤除菌。 |
| Elution Buffer | 1L | 50 mM NaH2PO4 (7.80 g NaH2PO4·2H2O) 300 mM NaCl (17.54 g NaCl) 250 mM imidazole (17.0 g imidazole) 使用NaOH溶液调节pH至8.0, 使用0.22 或者0.45 μm滤膜过滤除菌。 |
| 名称 | 体积 | 配方 |
| Lysis Buffer | 1L | 8 M Urea (480.50 g urea) 100 mM NaH2PO4 (15.60 g NaH2PO4·2H2O) 100 mM Tris·Cl (15.76 g Tris·HCl) 使用盐酸溶液调节pH至8.0 使用0.22 或者0.45 μm滤膜过滤除菌。 |
| Wash Buffer | 1L | 8 M Urea (480.50 g urea) 100 mM NaH2PO4 (15.60 g NaH2PO4·2H2O) 100 mM Tris·Cl (15.76 g Tris·HCl) 使用盐酸溶液调节pH至6.3 使用0.22 或者0.45 μm滤膜过滤除菌。 |
| Elution Buffer | 1L | 8 M Urea (480.50 g urea) 100 mM NaH2PO4 (15.60 g NaH2PO4·2H2O) 100 mM Tris·Cl (15.76 g Tris·HCl) 使用盐酸溶液调节pH至4.5 使用0.22 或者0.45 μm滤膜过滤除菌。 |
| 名称 | 体积 | 配方 |
| Lysis Buffer | 1L | 8 M Urea (480.50 g urea) 50 mM NaH2PO4 (7.80 g NaH2PO4·2H2O) 300 mM NaCl (17.54 g NaCl) 10 mM imidazole (0.68 g imidazole) 使用NaOH溶液调节pH至8.0, 使用0.22 或者0.45 μm滤膜过滤除菌。 |
| Wash Buffer | 1L | 8 M Urea (480.50 g urea) 50 mM NaH2PO4 (7.80 g NaH2PO4·2H2O) 300 mM NaCl (17.54 g NaCl) 20 mM imidazole(1.36 g imidazole) 使用NaOH溶液调节pH至8.0, 使用0.22 或者0.45 μm滤膜过滤除菌。 |
| Elution Buffer | 1L | 8 M Urea (480.50 g urea) 50 mM NaH2PO4 (7.80 g NaH2PO4·2H2O) 300 mM NaCl (17.54 g NaCl) 250 mM imidazole (17.0 g imidazole) 使用NaOH溶液调节pH至8.0, 使用0.22 或者0.45 μm滤膜过滤除菌。 |
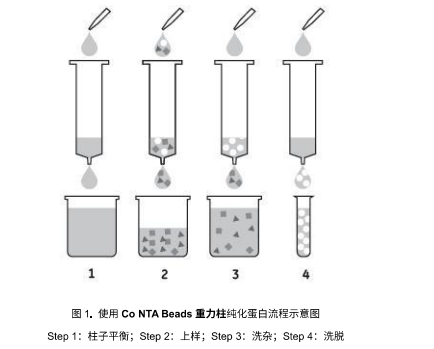
| 问题 | 原因分析 | 推荐解决方案 |
| 柱子反压过高 | 填料被堵塞 | 裂解液中含有微小的固体颗粒,建议上柱前使用滤膜(0.22 or 0.45 μm)过滤,或者离心去除。 |
| 样品太粘稠 | 样品中含有高浓度的核酸,加长破碎时间直至粘度降低,或者添加DNase I (终浓度5 μg/ml),Mg2+ (终浓度 1 mM),冰上孵育10-15分钟。 | |
| 缓冲液太粘稠 | 有机溶剂或者蛋白稳定试剂(如甘油等)可能会引起反压增高,降低操作流速。 | |
| 洗脱组分中没有目的蛋白 | 蛋白可能是包涵体,没有在上清中 | 可以通过电泳检测裂解液分析上清中是否含有目的蛋白,包涵体蛋白需要按照包涵体蛋白的纯化方式。 |
| 表达量太低 | 优化表达条件。 | |
| 目的蛋白结合比较弱,在洗杂步骤被洗下来了 | 提高wash Buffer的pH,或者降低咪唑浓度。 | |
| 目的蛋白结合过强,不容易洗脱下来 | 降低Elution Buffer的pH值,或者增加Elution Buffer中咪唑浓度。 | |
| 使用10-100mM EDTA溶液剥离钴离子,同时得到目的蛋白。 | ||
| 蛋白降解 | 在4°C下进行纯化操作。 | |
| 菌体破碎时添加一些蛋白酶抑制剂。 | ||
| 洗脱组分不纯(含有多种蛋白) | 洗杂不彻底 | 增加Wash Buffer体积。 |
| 样品中含有其他的组氨酸标签蛋白 | 通过调节pH值,或者咪唑浓度来优化洗杂条件,再使用其他的纯化手段(如离子交换,疏水等)进一步纯化洗脱组分。 | |
| 填料颜色变浅或变成白色 | 钴离子脱落或被剥离 | 按照第三部分填料再生中的建议重新挂钴离子 |
| 上样过程中蛋白发生沉淀 | 操作温度太高 | 4℃下进行上样。 |
| 蛋白发生聚集 | 在样品和所有的缓冲液中添加稳定剂,如0.1%的Triton X-100 或者Tween-20。 |
 电话咨询
电话咨询
 在线咨询
在线咨询
 QQ
QQ
 二维码
二维码
 扫码二维码
扫码二维码












